Research
Drug Discovery
Intrinsically disordered proteins
Many proteins have been found to be without stable structure in their native states. They are called intrinsically disordered proteins. Their ubiquitous presence undercuts the principle that a protein’s structure determines its function. However, the origins of disorder and its role in protein’s function are still not well understood. Our comparative genomic analysis showed that proteins' propensity for disorder depends strongly on their function and organism. Our thermodynamic analysis further showed that evolution may act differentially upon the level of disorder to optimize protein function. (more)
Protein - Protein interactions
Over the years we have developed a series of powerful methods to predict protein-protein interactions. One successful application involved the prediction and validation of a SUMOylated - Actin complex. Currently, we are interested in understanding allostery in the regulation of protein interactions.
Protein - DNA interactions
There is an urgent need to improve the predictive power of physically-based empirical potentials in order to better understand the molecular basis of protein function and expedite molecular engineering and drug design. Traditional molecular dynamics techniques can compute free energy differences between well-defined states, but they cannot consistently estimate the full contribution of the solvent. As a result, considerable effort is being devoted to the development of polarizable force fields. Despite their solid theoretical foundation, polarizable force fields have yet to reach their full potential and, equally important, justify their computational cost. My lab is working in developing a scalable approach for representing the polarization effect of molecular water by using a novel structure-based method to estimate physical interactions. The main hypothesis is that varying dielectric environments can be modeled using a "water factor" derived from a structural analysis of molecular water in crystal structures. Our approach goes beyond solvent accessible surface area techniques: both the accessibility to solvent and the nature of the water network ("bulk", "trapped", or "crystal") are taken into account.We have used this insight in the development of a novel optimization framework, based on mixed integer programming, that automatically derives empirical binding models and interaction parameters from high-quality binding data and crystal structures. As a proof-of-concept, we have used our technology to derive an interaction code for interactions between DNA and C2H2 zinc finger transcription factors. Our method improves the R2 correlation between predicted and experimental changes in binding free energy from less than 0.7 for previous approaches to 0.93—0.99 for different zinc-finger classes. This improvement is due almost exclusively to the application of our "water factor" insight. Our novel mathematical framework can efficiently explore a large space of probable interaction models and produce an optimal set of interaction parameters.
Interactions between Protein and Disordered Peptides.
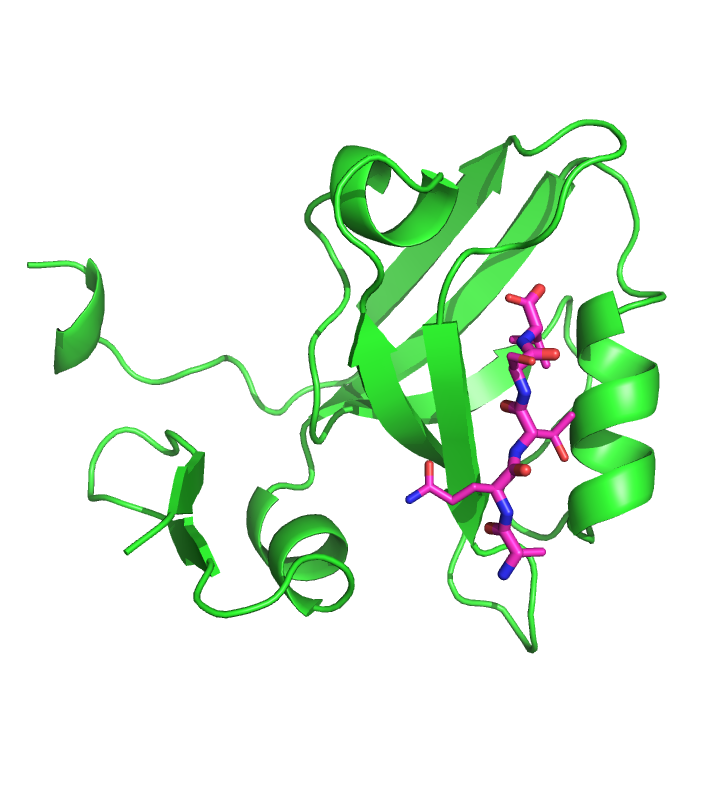
Crystal structure of PSD95-PDZ3 with CRIPT peptide. (PDB: 1BE9)
PDZ domains bind the disordered C-terminus of plasma membranes, mediating protein-protein interactions. These domains are highly promiscuous, and characterizing their binding specificity is critical to reveal their multiple roles in signal transduction. Crystallographic studies have revealed that binding PDZ scaffolds requires a four residue long strand anchored by a C-terminal hydrophobic residue. Based solely on the recognition motif of one co-crystal, we developed the first semi-flexible docking method to predict both the bound structure and affinity of disordered peptides with PDZ domains. A binding threshold of 10-5 M leads to sensitivity-specificity rates of 80%-80% in two independent datasets of 126 and 95 10-residue long natural and artificial peptides screened against the third PDZ domain of PSD95. Moreover, five peptides bound to 4 different PDZ domains are confirmed to form specific interactions, and their bound models are accurately predicted as the most stable complexes...(more)