Welcome to Carlos J Camacho's Lab
Research
Intrinsically Disordered Proteins
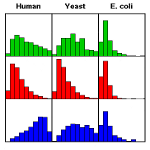
Intrinsic protein disorder has been found in many proteins. However, its origin has not been well understood. Our genome-wide survey indicates that the distribution of disorder depends strongly on protein function, and a first principles thermodynamic analysis explains the nature of this relationship. (more)
Protein-Protein Interactions
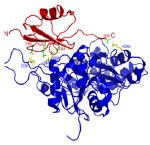
We have developed a fast algorithm for filtering docked conformations with good surface complementarity, and ranking them based on their clustering properties. The free energy filters select complexes with lowest desolvation and electrostatic energies.
Protein-DNA Interactions
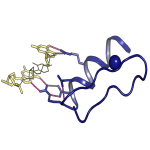
We developed a novel experimentally based approach to accurately and quantitatively estimate inter-molecular contact free energies and atomic desolvations in protein and DNA interactions. The method provides a general framework to design and optimize EGR-like transcription factors in silico.
Protein-Peptide Interactions
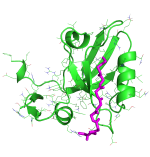
We use PDZ domain as a model, trying to understand what's the binding mechanism of the interactions between protein and disordered peptides. We successfully predicted complex structure and discriminate strong binders from weak binders.
Peptide Immunogenicity
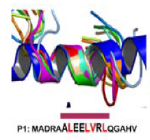
Using thermodynamic modeling and MD simulations, this work provides a framework for understanding the relative capacity of inherently unstable peptide structures to faithfully trigger B cell antibody production against specific conformational motifs found in native/intact proteins.
Drug Development
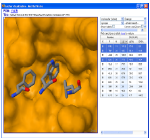
We are developing tools that rapidly search MCR chemical space for novel protein-protein inhibitors. We automatically identify anchors, amino acids critical to binding, and perform interactive pharmacophore searches of anchor-oriented compounds. We are actively investigating several targets using this approach and are developing methods to expand our search capabilities and refine the results of our pharmacophore search.